BiteSized Immunology: Immune Dysfunction

Angioedema due to acquired C1-Inhibitor deficiency
Introduction
Acquired angioedema (AAE) due to deficiency of C1-inhibitor is a relatively infrequently occurring but serious disorder, resulting in severe, sometimes life-threatening, episodes of angioedema. The precise incidence is unknown.
Clinically, angioedema caused by an acquired deficiency of C1-inhibitor is indistinguishable from hereditary angio-edema (HAE). However, the condition usually occurs in adults over 40 years of age and there is no familial predisposition. Angioedema typically affects the face, the oropharyngeal and laryngeal region (which may cause airway obstruction), the mesenteric and intestinal site (causing severe abdominal pain, diarrhea and vomiting), the genital region, or extremities. Physical or psychosocial stress are recognized as provoking factors for an angioedema attack, however, in many cases, no clear triggers can be identified.
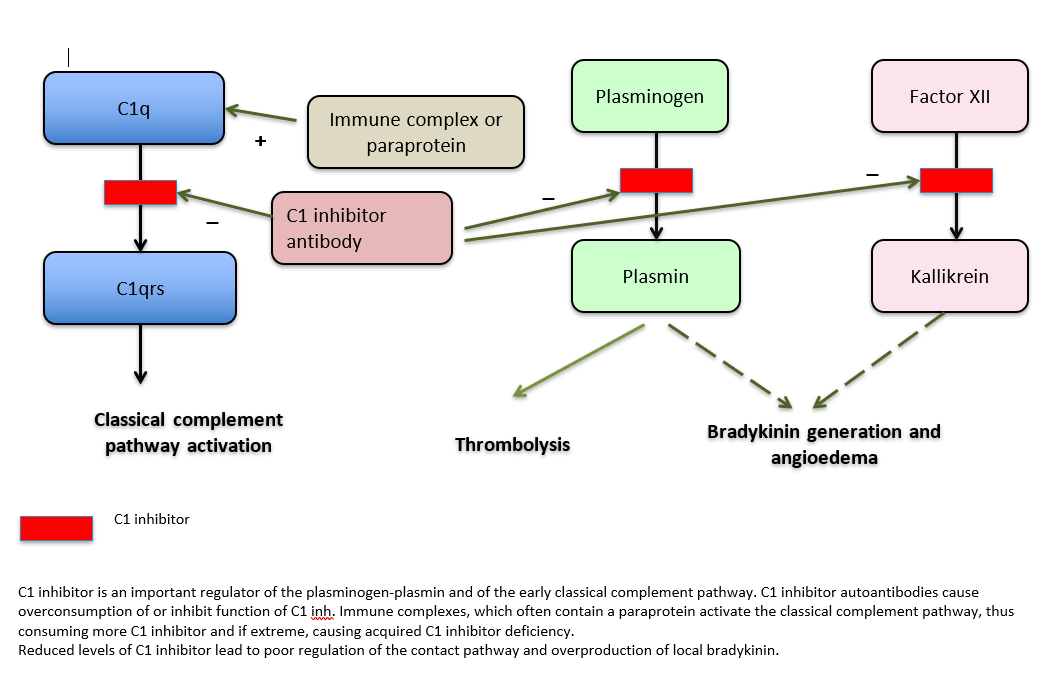
Acquired C1-inhibitor deficiency and B-cell disorders
The main causes of acquired C1-inhibitor deficiency are lymphoproliferative disease and autoimmune disease. Most cases of acquired C1-inhibitor deficiency are associated with an underlying B-cell disorder, ranging from auto-immune anti-C1-inhibitor auto-antibodies to lymphoproliferative disease (usually a low grade or splenic marginal zone lymphoma). There are two mechanisms which can lead to this condition: consumption due to complement activation, or (rarely) due to an autoantibody directed against C1-inhibitor. Interestingly, many patients with lymphoproliferative disorders may have anti-C1 inhibitor antibodies but without any clinical evidence of angioedema. The clinical manifestations of angio-edema may precede the diagnosis of a lymphoproliferative disorder in some cases by many years.
Diagnosis
The diagnosis of acquired angio-edema is based on a (very) low level of C1-inhibitor concentration and its function and (usually) low complement factor 4 (C4) levels. In contrast to hereditary angio-edema, plasma levels of C1q are decreased in AAE which sometimes helps with the differential diagnosis. Anti-C1 inhibitor antibodies can be detected in some but not all cases. If there is doubt whether the deficiency of C1 inhibitor is hereditary or acquired, genetic analysis of the SERPING1 gene can exclude or confirm any pathogenic mutations associated with HAE. In cases where there is a strong suggestion the angioedema is hereditary genetic analysis may find mutations in genes for factor XII, plasminogen or angiopoietin-1.
Most clinicians will screen for a lymphoproliferative disorder in patients with acquired angio-edema by physical examination and looking for lymphoadenopathy, performing a CT chest and abdomen as well as assessing a full blood cell count, serum immunoglobulins and paraprotein.
Treatment
Patients with acquired angioedema are managed by treating the underlying disorder with a clinical benefit in most cases. Patients in whom no such underlying disease has been established, are initially treated with prophylactic lysine analogues, such as tranexamic acid (which acts by inhibition of plasmin formation), that plays a crucial role in angioedema development. Androgenic steroids, such as danazol (which increases endogenous hepatic synthesis of C1-inhibitor and aminopeptidase P involved in bradykinin degradation) is helpful in some cases. In case of severe angioedema (for example located in the oropharyngeal and laryngeal areas or serious abdominal attacks), administration of C1-inhibitor concentrate (either plasma-derived or recombinant) or of the bradykinin receptor B2 antagonist icatibant is indicated. Administration of immunosuppressive drugs in patients with anti-C1 inhibitor autoantibodies may sometimes be clinically efficacious.
As the underlying causes of acquired C1 inhibitor deficiency in most, if not all, patients are related to B-cell dysfunction, anti-CD20 targeted treatments are thought to be a rational strategy. There are several case-reports confirming the efficacy of rituximab for the treatment of acquired angioedema due to C1-inhibitor deficiency. From the published reports it seems that rituximab is effective in both auto-antibody associated C1-inhibitor deficiency as well as C1-inhibitor deficiency associated with a lymphoproliferative disorder. A recent report demonstrated the long-term remission of patients with acquired C1-inhibitor deficiency and severe angioedema treated with rituximab (Levi M et al, Long-term effects upon rituximab treatment of acquired angioedema due to C1-inhibitor deficiency. Allergy. 2018 Nov 29. doi: 10.1111/all.13686.
Further research
The efficacy and safety of new agents used for the treatment of hereditary angio-edema (such as oral or subcutaneous kallikrein inhibitors) needs to be investigated and established in patients with acquired angio-edema due to C1-inhibitor deficiency.
A better understanding of the pathogenesis of lymphoproliferative disorders causing acquired C1-inhibtor deficiency may lead to more targeted and effective treatment modalities.