BiteSized Immunology: Pathogens & Disease

Host − Pathogen interactions and immune evasion
Any microorganism which is able to cause disease in a host organism is termed a pathogen. This article is confined to human microbial pathogens, although plant and animal pathogens are also widespread in nature. When a pathogenic microorganism (bacterium, virus or protozoal parasite) infects the human body, a battle ensues between the host’s innate & adaptive immune systems and the pathogen’s assorted virulence mechanisms and factors. The outcome of this battle determines whether, and how well, the host survives and recovers. Full recovery entails the achievement of physiological (and immunological) homeostasis in the host, and the length of time this takes will depend on the nature and severity of the infection and whether there has been any prophylactic or therapeutic intervention. Many pathogens also deploy diverse immune evasion tactics in the host to achieve host cell invasion and colonisation and may successfully exploit host cells to access target tissues.
This article presents a selection of both virulence and immune evasion strategies, the latter of which may involve: (1) hiding from the immune system (e.g. within cells); (2) interfering with the function of the immune system (e.g. blocking signals); (3) destroying elements of the immune system (e.g. the structures which present microbial antigens to immune effectors to initiate a response in the host). Virulence generally involves the employment of various mechanisms to destroy, or cause the malfunction of, host cells. The host may also employ counterstrategies in response to these. A more detailed description of the various mechanisms employed by specific microorganisms can be found on other Bite-sized pages.
Viruses such as Varicella zoster (chickenpox) and Herpesviridae (herpes simplex viruses, Varicella-Zoster virus, cytomegalovirus etc) can hide from the immune system in neurons and non-neuronal cells where they may persist for many years, before emerging in pathogenic form when the host has a lowered resistance. This is the case also for bacteria such as Borrelia burgdorferi and Burkholderia pseudomallei (causative of Lyme disease and melioidosis, respectively) where there are reports that symptoms of infection have re-emerged months to years (B.burgdorferi), and even up to 60 years (B.pseudomallei), after the initial infection. In terms of immune interference, more overt strategies may be deployed, e.g. Leishmania protozoal parasites belonging to Leishmania spp. can selectively inhibit the transcription of the pro-inflammatory cytokine interleukin 12 (IL12p40) in the host, thus suppressing the host’s immune response.
To sustain their virulence mechanisms, many bacteria can sequester free iron in the mammalian host, through the elaboration of iron-binding siderophores. Iron is an essential component of metabolism in both the host and the micro-organism. Thus to protect itself from such virulence mechanisms, the host cell fights back by synthesising siderocalin receptors which competitively bind iron. Mammalian host cells have also evolved an array of pattern recognition receptors for microbes or microbial factors, such as the Toll-like receptors (TLRs), which when bound, trigger intracellular signalling cascade(s) with antimicrobial effects.
Whilst many bacterial pathogens are intracellular in nature, others do not need to invade the host cell, but instead use various secretion processes which effect the delivery of toxins and other virulence factors into the host cell. Examples of bacteria which have developed the ablity to make a hollow projection (a so-called translocon) which on contact with the host cell can deliver anti-host factors into it, often resulting in host cell apoptosis (so-called Type III secretion), include Escherichia coli, Shigella flexnerii, Yersinia pestis and Chlamydia trachomatis which cause diverse syndromes of food poisoning, dysentery, bubonic plague and genito-urinary tract infection, respectively (see Figure 1). However, some bacteria such as Francisella tularensis (causative of tularemia) and Burkholderia spp. (causative of melioidosis or glanders) have multiple secretion processes through which they deliver virulence factors into the host cell.
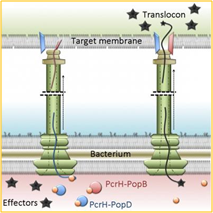
Another serious human pathogen, Bacillus anthracis, causative of anthrax, has well-developed virulence mechanisms involving the secretion of three proteins, one of which, protective antigen (PA), binds host cell receptors to effect entry of either lethal factor (LF) or edema factor (EF). The PA−LF or PA−EF complexes undergo clathrin-mediated endocytosis and enter early endosomes with subsequent microtubular transport through vesicles into acidified late perinuclear endosomes. Under these conditions, LF is released into the cytoplasm, whereas EF remains bound to the late endosomal perinuclear membrane. In the cytoplasm, LF cleaves and inactivates mitogen-activated protein kinase kinases (MAPKKs) to disrupt phosphorylation and transcription in the nucleus, ultimately preventing protein synthesis and causing cell death; whilst EF, a calcium and calmodulin-dependent adenylate cyclase, causes a rapid increase in perinuclear cAMP resulting in cellular, tissue and ultimately organ edema. Both LF and EF also have the effect of suppressing pro-inflammatory cytokine secretion and weakening vascular endothelial barriers by downregulating vascular cadherin, which is important in cell−cell adhesion; these effects contribute to the vascular leakage typical of systemic anthrax.
Viral pathogens, on the other hand, do need to invade a host cell to complete their replication cycles. One of the most serious pathogens to emerge in recent years, human immunodeficiency virus (HIV), hijacks CD4+ T-cells to degrade the host’s ability to retaliate with a strong cell-mediated immune (CMI) response. The immune evasion tactics used by HIV are so far-reaching that they have so far stymied progress towards the advanced development of an effective vaccine. Firstly, HIV gains access to a naive subject in immune-privileged sites, such as the vagina or rectum, which are not well supplied with lymphoid tissue; subsequently the virus utilises co-receptors (CCR5/CXCr4) to gain access to CD4+ host cells. The outcome is a gradual loss of CD4+ T cells with attrition of CMI function in the host and an increased susceptibility of the host to other infections (bacterial pneumonia) or to tumours (e.g. Kaposi’s sarcoma). HIV cases are controlled by anti-retroviral drugs, whilst vaccine candidates are being identified and evaluated. Variola major (causative of smallpox) has also evolved host immune evasion tactics, by secreting a protein which inhibits the activation of complement enzymes and another, chemokine-binding protein, type II, which blocks signals calling for immune cells and inflammation at the infection site. Pathogens may also evade the host’s antigen processing and presentation system e.g. by interfering with the expression of surface MHC class I proteins, although this in turn increases their susceptibility to Natural Killer (NK) cell activity. However, members of the herpesvirus, papillomavirus, retrovirus, poxvirus and flavivirus families have also evolved mechanisms to escape NK cell attack and to promote their own survival in host cells by inhibiting host cell apoptosis. Finally, some pathogens frequently change the surface antigens they display, the influenza virus (orthomyxovirus) being a prime example of this.
Through all these virulence mechanisms and immune evasion techniques, pathogens have evolved strategies to survive in the host. Of course, the interaction of a pathogen with a host is a dynamic situation, so that advances made by the pathogen are countered by the host. Thus, the expansion of cytotoxic T cells (Tc) in the host can lead to escape mutations in the pathogen to avoid being a future target for Tc. Hence pathogens will continue to evolve and emerge, and arguably the most successful are those that exploit their hosts without killing them.
© The copyright for this work resides with the author.