BiteSized Immunology: Immune Dysfunction

Type 1 Diabetes
Type 1 diabetes (T1D) is a chronic T-cell mediated disease that leads to the destruction of the insulin-secreting islet β-cells (Figure 1) resulting in absolute insulin deficiency and hyperglycaemia.
Epidemiology
Type 1 diabetes (T1D) accounts for about 5-10% of all patients with diabetes and the worldwide incidence is increasing by ~3 % every year. There is considerable geographical variation in the prevalence of the disease with the highest incidence seen in Finland and Sardinia and the lowest in Venezuela.
Pathology
The clinical presentation of T1D is preceded by an asymptomatic period lasting from months to years which is characterised by autoantibodies against islet β-cell components. Insulin autoantibodies are amongst the first to appear, followed by autoantibodies to glutamic acid decarboxylase (GAD) and then spreading to IA-2 (insulinoma-associated tyrosine phosphatase protein) and ZnT8 (zinc-transporter 8); indeed, the presence of multiple autoantibodies is diagnostic of stage 1 of T1D (Fig 1).The destruction of the islet β-cell in T1D is the result of a complex interplay between multiple players of both the innate and adaptive immune system; immunohistochemical analysis of islet inflammation from pancreata of patients with T1D obtained at autopsy indicate a mononuclear cell infiltrate in islets (termed insulitis) consisting mainly of macrophages, B cells and T cells. Both CD4+ and CD8+ T cells are required for disease development, by destroying the insulin-producing β cells through the effector functions of Th1 cells and direct killing by cytotoxic T lymphocytes (CTLs). CTLs initiate killing by various mechanisms including the production of inflammatory cytokines such as TNF-a and IFN-g, which act synergistically with IL-1 produced by macrophages in targeting the β-cells; they also directly kill β-cells through the secretion of perforin or by apoptosis by the activation of the Fas-Fas-L pathway.
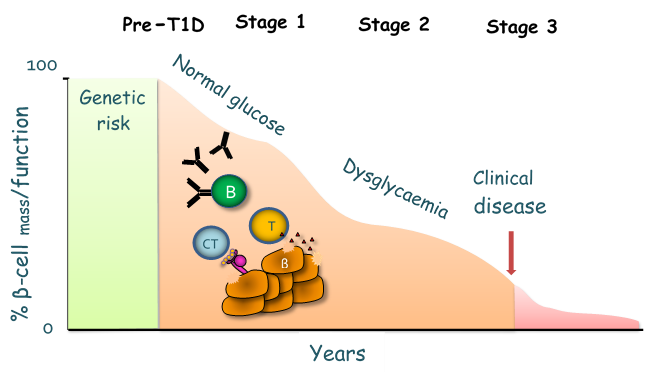

The destruction of the islet β-cell in T1D is the result of a complex interplay between multiple players of both the innate and adaptive immune system; immunohistochemical analysis of islet inflammation from pancreata of patients with T1D obtained at autopsy indicate a mononuclear cell infiltrate in islets (termed insulitis) consisting mainly of macrophages, B cells and T cells. Both CD4+ and CD8+ T cells are required for disease development, by destroying the insulin-producing β cells through the effector functions of Th1 cells and direct killing by cytotoxic T lymphocytes (CTLs) (Fig 2). CTLs initiate killing by various mechanisms including the production of inflammatory cytokines such as TNF-α and IFN-g, which act synergistically with IL-1β produced by macrophages in targeting the β-cells; they also directly kill β-cells through the secretion of perforin or by apoptosis by the activation of the Fas-Fas-L pathway.
The rate and extent of β-cell destruction can vary amongst patients, as can the age of onset and the number and type of autoantibodies present at diagnosis thus T1D is heterogenous in presentation.