BiteSized Immunology: Pathogens & Disease

Microbial infection in cystic fibrosis
Cystic fibrosis
Cystic fibrosis (CF) is the UK’s most common inherited disease affecting around 1 in 2,500 births (predominantly affecting Caucasians). It is an autosomal recessive disease, i.e. the faulty gene occurs on an autosomal chromosome and two copies of the defective gene are required to develop the condition. In the UK, around 2 million people are carriers and although they do not have the disease, two carriers have a 1 in 4 chance of having a child with CF. The defective gene is the cystic fibrosis transmembrane conductance regulator (CFTR). The CFTR protein is present on epithelial cells throughout the body. It is a chloride ion channel involved in maintaining the water and ion homeostasis on cell surfaces. As it is ubiquitously expressed, multiple organs are affected. Initially infant CF patients have numerous digestive problems, with over 90% of patients suffering from pancreatic disease (insufficiency) which may lead in later life to the development of diabetes. Pancreatic insufficiency causes malabsorption which correlates with poor growth and weight gain. Reproduction is also affected, more so in men with 98% infertile, but not sterile, although able to have children with assisted fertility techniques). However female fertility may be impaired due to dehydration of the cervical mucus, but reproductive function still remains normal. Despite the various complications linked to the disease, the main cause of morbidity and mortality in CF is lung disease. It is the main characteristic feature of CF and is a result of an exaggerated pro-inflammatory response following bacterial infection.
CFTR and lung disease
To date over 1,500 mutations of the CFTR protein are known. The F508del mutation accounts for 50-70% of cases worldwide (90% in the USA). F508del arises from a deletion of phenylalanine at position 508 on the CFTR protein, resulting in a misfolded protein that fails to translocate to the apical membrane. The lungs are lined with epithelial cells containing the CFTR on the apical surface. Through the movement of chloride ions, CFTR facilitates hydration of the airway surface liquids (ASL). The ASL contains a liquid layer in which the cell cilia bath, called the periciliary layer, as well as a mucus layer, which traps invading bacteria. In non-CF lungs, the secretion of chloride onto the ASL draws water from the epithelial cells, hydrating the ASL layers. Bacteria are efficiently swept away with the mucus through cilia movement (Figure 1). In CF lungs hydration of the ASL is diminished, resulting in think and sticky mucus, which provides the perfect environment for bacteria to infect and propagate. In the less hydrated periciliary layer, the cilia are flattened and the ability to clear bacterial infection reduced.
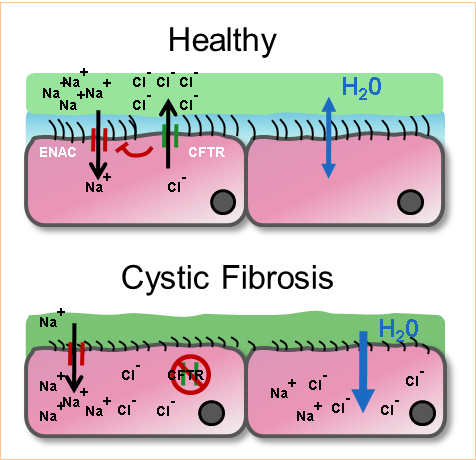
Bacterial Infection
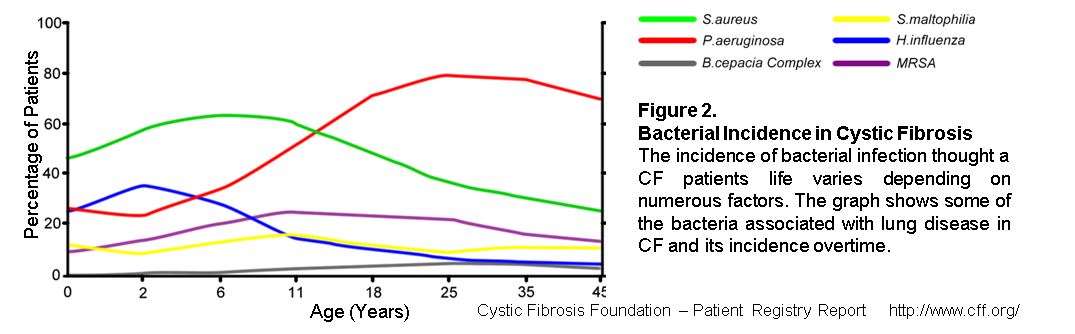
Patients with CF experience multiple bacterial infections throughout life, however the bacterial flora in the lungs is subject to change over time (Figure 2). Initially the main colonising bacteria is the Gram-positive Staphylococcus aureus, which affects around 50% of patients during infancy. However, its incidence declines later in life and infections with the Gram-negative Pseudomonas aeruginosa become more prominent and by the age of eighteen around 80% of patients are chronically infected with the bacteria. Other bacteria also known to infect the CF airways include H. influenza, MRSA and S. maltophillia. While infection with these bacteria can be routinely identified and the appropriate treatment given, other bacteria such as Burkholderia (ceno)cepacia are harder to detect and treat. Infections with B. (ceno)cepaecia have a low incidence, but is associated with a high mortality rate.
How does the immune system detect bacterial infection?
Bacterial products and components are collectively named pathogen associated molecular patterns (PAMPs) and are detected by pattern recognition receptors (PRRs) expressed by cells of the immune system. The detection of PAMPs by PRRs and their subsequent activation results in an innate and acquired inflammatory immune response. One such family of PRRs is the Toll-like receptor (TLR) family. The different TLRs are characterised by their ligand specificity, some detecting extracellular bacteria or bacterial products, others are located and function intracellularly.
Toll-like receptors (TLR) and lipopolysaccharide (LPS)
Currently there are 10 known human TLRs which bind various PAMPs from numerous microorganisms. For example, TLR-2 can bind peptidoglycan, lipoteichoic acid and lipoproteins, all of which are found on the membrane of Gram-positive bacteria such as S. aureus. Flagellin binds to TLR-5 and bacterial RNA and DNA is detected by TLR-3 and TLR-9, respectively. However, one of the most studied PAMPs is lipopolysaccharide (LPS), the main cell wall component of Gram-negative bacteria such as P.aeruginosa. Binding of LPS to TLR-4 results in activation of nuclear transcription factors and subsequently the release of pro-inflammatory cytokines.
The PAMPs mentioned above are commonly found throughout the airways of CF patients and stimulate the activation of PRR resulting in the commonly seen inflammation in CF. The exaggerated response that results from bacterial infection causes the production of cytokines such as interleukin (IL)-6, IL-8 and TNF-a. This results in a massive influx of neutrophils into the airways, which contribute further to the damage caused by inflammation.
Treatment
Treatment and care to improve quality of life plays a major role the life a person with CF. Pancreatic enzymes will combat pancreatic insufficiency and will help to prevent malnutrition. Nutritional supplements are also given. The main area of treatment is directed at the respiratory symptoms. Clearance of the accumulated mucus that is colonised with bacteria is crucial to reduce lung inflammation and improve lung function. This care is often given by a physiotherapist and includes postural drainage and clapping, but aerobic exercise also improves clearance.
Cell damage and bacterial death within the airway surface liquid (ASL) increases the DNA content in the mucus. DNase is a relatively new treatment used to cleave free DNA into smaller fragments. This reduces mucus viscosity and aids clearance. However, the main treatment in CF over the past 60 years has been the use of culture-specific antibiotics to clear bacterial infection, therefore reduce tissue damage from inflammation and subsequently slow down the accelerated decline in lung function.
Survival and the future
With improvements in antibiotic treatments and the development of new drugs the life expectancy of CF patients has increased from just 25 years in 1980 to nearly 40 years at present. Currently over 35% of CF patients are over the age of 18 years old, a figure that continues to increase every year. Recently a lot of intensive research has gone into gene therapy and CFTR correctors, in order to rescue the CFTR from the endoplasmic reticulum to the membrane and help restore normal function in the lungs. However, as long as these strategies have not been completely successful, the focus remains on radical antibiotic treatment that can control bacterial infection and inflammation. Reducing lung inflammation may help to reduce lung damage and increase lung function. Overall, further research in all of the areas outlined above are needed to drive the production of new treatments and improvement in care in a bid to further increase life expectancy and hopefully finally cure CF.
© The copyright for this work resides with the author.